1 主題內容與適用范圍
本標準規定了以活性炭為主要吸附劑,以去除有機物為目的的飲水凈器(以下簡稱凈水器)的技術要求,試驗方法和檢驗規則。
本標準適用于以自來水為進水的供家庭和集團飲用的活性炭凈水器。
2 引用標準
GB 5749 生活飲用水衛生標準
GB 5750 生活飲用水標準檢驗法
GB 8538 飲用天然礦泉水檢驗方法
GB 4804 搪瓷食具容器衛生標準
搪瓷食具衛生管理辦法
GB 2633 日用搪瓷制品檢驗方法
GB 4803 食品包裝用聚氯乙烯樹脂衛生標準
食品用塑料制品及原材料衛生管理辦法
GB 7702 煤質顆粒活性炭—有效防護時間測定總方法
GB 601 標準溶液制備方法
GBn 84 聚乙烯成型品衛生標準
GBn 85 聚丙烯成型品衛生標準
GBn 86 聚苯乙烯成型品衛生標準
GB 9684 不銹鋼食具容器衛生標準
GB 4807 食用橡膠墊片(圈)衛生標準
GB 4805 食品罐頭內壁環氧酚醛涂料衛生標準
QJ 1289 結構鋼、不銹鋼電阻點,縫焊技術條件
3 術語
3.1 集團用 For drinking purpose of groups or ouganizations
指供學校、廠礦、機關、賓館等團體使用。
3.2 產水率 Rate of out flow
每分鐘經凈水器產出的水量,以L/min計。
3.3 額定總產水量 Rated total output capacity
凈水器中活性炭的吸附容量所能承受的產水量總和,L(家庭用)或m3(集團用)。
3.4 相對產水率 Relative rate of out flow
以活性炭體積倍數計算的凈水器每分鐘的產水量即空間流速以min-1計。
3.5 相對總產水量 Relative total output capacity
以活性炭體積的倍數計算的凈水器累計產水總量。
4 產品分類
4.1 產品品種
家庭用凈水器和集團用凈水器。
4.2 產品型號與標記
產品標記由產品名稱,品種代號,質量等級代號,產水率代號和額定總產水量代號組成。
××× ×× ×× × ×
其中:×××——活性炭凈水器;
××——家庭用或集團用;
××——質量等級;
×——產水率;
×——額定總產水量。
4.2.1 家庭用凈水器的代號為H,集團凈水器代號為G。
4.2.2 質量等級的代表為A或B。A表示一級品,B表示二級品。
[例1]家庭用活性炭凈水器,裝活性炭1L,一級品,產水率1.0Lmin,額定總產水量2000L,產品型號及標記為:
活性炭凈水器 HA 1.0 200
[例2]集團用活性炭凈水器,裝活性炭體積0.2m3,二級品,出水率10L/min,額定總產水量200m3。產品型號及標記為:
活性炭凈水器 GB 10 200
5 技術要求
5.1 材料要求
5.1.1 與水接觸的凈水器殼體和管道采用的搪瓷、塑料或不銹鋼材料應分別符合GB4804、GB4803、GB9684、GBn9684、GBn85、GBn86的規定。
5.1.2 用以截留懸浮物的纖維網布或多孔濾膜,濾片和濾柱應是食品級材料制成。或經當地衛生監督部門檢驗核準的材料。
5.1.3 凈水器所用橡膠墊片(圈)的衛生要求應符合國際GB4807的規定。
5.1.4 凈水器內壁涂料應符合GB4805的規定。
5.1.5 凈水器采用的顆粒活性炭應符合表1的要求:
表1 顆粒活性炭技術要求
| 項 目 | 指 標 | | A級凈水器用 | B級凈水器用 | | 水 份(%) | ≤5 | ≤5 | | 強 度(%)(球磨法) | ≥90 | ≥85 | | 碘吸附值(mg/g) | ≥1000 | ≥800 | | 亞甲蘭吸附值(mg/g) | ≥135 | ≥105 | | 苯酚吸附值(mg/g) | ≥120 | ≥120 | | 半脫氯值(cm) | ≤6.0 | ≤8.0 | | 堆集密度(g/cm3) | ≤0.5 | ≤0.5 | | 粒 度 | 不規定 | | pH | 4~11 | | 氯化物 (%) | ≤0.5 | | 鉛 (µg/g) | ≤10 | | 鋅 (µg/g) | ≤50 | | 鎘 (µg/g) | ≤1 | | 砷 (µg/g) | ≤2 | 5.2 制作要求
5.2.1 凈水器活性炭過濾柱的有效高度與直徑之比采用2~6為宜。
5.2.2 管道、管件要求應橫平、豎直,法蘭要求應上下平行。
5.2.3 過水容器應在規定試驗壓力下無滲漏、損壞。
5.2.4 搪瓷筒體應按GB2633方法檢驗,其密著性能試驗不得呈塊狀;耐熱驟變試驗不得掉瓷;耐堿試驗不得失去原有的光澤;耐酸試驗侵蝕≤0.2mg/mL。
5.2.5 塑料筒體對稱部位壁厚比不應大于3:2,嘴口要求應平整,廢邊應整修光滑,螺紋清晰,配合適宜。
5.2.6 不銹鋼筒體的焊接應符合QJ1289的要求。
5.3 裝配要求
5.3.1 與水接觸的所有零部件必須經清洗和消毒。
5.3.2 活性炭在放入容器前應先裝入尼龍篩絹網,并進行清洗消毒和干燥處理。
5.3.3 凈水器各零部件的裝配應保證無松動現象發生,并便于更換活性炭。
5.4 外觀要求
5.4.1 搪瓷筒體外表應符合GB4804的要求。
5.4.2 塑料筒體外表不應有砂眼或塑化不良,應光滑平整,不應夾帶雜質或表面凹凸等明顯有損外觀的缺陷。
5.4.3 不銹鋼筒體外表應光亮,不應有明顯影響外觀的缺陷。
5.4.4 筒體外表應有明顯的產品標志。
5.5 凈水器的消毒設備要求
5.5.1 凈水器是否帶消毒設備應在產品說明書中予以說明。供生飲用的凈水器必須帶消毒設備。
5.5.2 凈水器的消毒可采用紫外燈,次氯酸鈉發生器,臭氧發生器,超濾膜過濾器或其他合適的消毒設備或措施。
5.5.3 消毒設備與措施應與凈水器的產水率及額定總產水量相適應,以保證凈水器出水水質符合GB5749中細菌學的要求。
5.6 使用性能要求
5.6.1 集團用凈水器及其附屬設備應保證在0.2MPa工作壓力下無泄漏,筒體不變形。家用凈水器應在0.1MPa工作壓力下無泄漏,筒體不變形。
5.6.2 凈水器及其附屬設備的產水率應保證在0.2MPa工作壓力下,不應小于標記產水率。凈水器的產水率及額定總產水量應根據活性炭的吸附容量確定。
5.6.3 凈水器在標記額定總產水量到達前,其出水水質應優于GB5749的要求,亞硝酸鹽氮應小于0.03mg/L。
5.6.4 當凈水器進水水質中個別有機物指標超過生活飲用水衛生標準限值時,凈水器對該指標在相對總產水量到達前應有一定的去除率。
5.6.5 采用三碘樹脂消毒的凈水器出水中含碘化物的濃度不得超過0.05mg/L。
5.6.6 進水耗氧量(高錳酸鉀指數)CODMn 為4.0mg/L,溶解性有機碳DOC為6.0mg/L時,在相對產水率的條件下運行的凈水器的凈水效率應符合表2要求。
6 試驗方法
6.1 生產廠應按5.1.1~5.1.5所列要求,提出有關的檢驗合格證書。
6.2 活性炭的質量應按GB7702規定的測定方法進行測定。
半脫氯值、pH、氯化物、砷、鋅、鎘、鉛的測定方法見附錄A,附錄B 。
6.3 5.2要求應按有關規定進行。
6.4 5.3~5.5要求按生產廠檢驗規范進行檢查。
6.5 應采用1.5級壓力表在封閉出水口的條件下進行凈水器的壓力試驗。
6.6 凈水器產水率在進水壓力為0.2MPa時,應用轉子流量計測定,或用秒表量筒測定。
表2 活性炭凈水器凈水效率 | 項 目 | 指 標 | A級產品相對產
水率(min-1) | B級產品相對產
水率(min-1) | | 1.0 | 0.2 | 1.0 | 0.2 | | 1.相對總產水量 | ≥1200 | ≥2400 | ≥600 | ≥1200 | 2.額定相對總產水量到達前出水
耗氧量CODMn瞬時去除率(%)
總溶解性有機碳DOC瞬時去除率(%) | ≥25
≥25 | ≥15
≥15 | 3.額定相對總產水量到達前
CODMn總去除量(g/L炭)
DOC總去除量(g/L炭) | ≥2.5 ≥5.0
≥4.0 ≥8.0 | ≥1.0 ≥2.0
≥1.5 ≥3.0 | 4.額定相對總產水量到達前出水的Ames致突變試驗
(1)進水為陽性時
(2)進水為陰性時 | 陰 性
陰 性 | 陰 性
陰 性 | 注:當進水的CODMn。DOC與上述指標不同時,應按進水的CODMn和DOC實際值折算相對總產水率。
6.7 家用凈水器應在相對產水率為1.0min-1時檢驗出水質;集團用凈水器應在相對產水率為0.2min-1時檢驗出水水質。產品的標記產水率與規定的相對產水率不一致時,應按大的產水率進行檢驗出水水質。
6.8 為檢驗凈水器的凈水效率,凈水率應按6.7規定的產水率,至少每天采進出水水樣測定CODMn及DOC或其他水質指標(視需要而定),直至總產水量超過相對產水率為止:或將采集的水樣等量混和后,測定混和水樣的CODMn及DOC或其他指標,并按表2中第1、第3項指標考核,但最后一次采集的水樣需單獨采集,并按表2的第2、第4項指標考核。
6.9 凈水器出水水質應按GB5750的檢驗法檢驗,磺化物檢驗應按GB8538分析方法檢驗。總溶解性有機碳DOC及Ames致突變的試驗方法見附錄C、附錄D。
7 檢驗規則
7.1 凈水器的產品檢驗分為出廠檢驗和型式檢驗。
7.2 出廠檢驗
7.2.1 凈水器在出廠前必須按規定的項目及試驗方法進行出廠檢驗,合格后方可出廠。
7.2.2 出廠檢驗項目
出廠檢驗項目包括本標準5.1~5.5的各項規定,用目視法解體檢查。
7.2.3 經出廠檢驗的產品數,家用凈水器應不少于出廠凈水器產品數的1/100;集團用凈水器應不少于出廠凈水器數的1/10。
7.3 型式檢驗
7.3.1 有下列情況之一時,應進行型式檢驗
a.新產品或老產品轉廠生產的試制定型鑒定;
b.正式生產后,如結構、材料、工藝有較大改變,可能影響產品性能時;
c.正常生產后,定期或積累一定產量后,(由當地技術監督部門規定)應周期性進行一次檢驗;
d.產品長期停產后,恢復生產時;
e.出廠檢驗結果與上次型式試驗有較大差異時;
f.國家質量監督機構提出進行型式檢驗要求時。
7.3.2 型式檢驗應按本標準第5章中各項技術要求和第6章規定的試驗方法進行。
7.3.3 型式檢驗應直接從市場隨機采樣,每一型號的產品,應采取2只試樣;一只按5.1~5.5的規定檢驗,一只按5.6的規定檢驗。
7.3.4 型式檢驗不合格的產品,應限期生產廠進行改進,到期復驗。
8 判定規則
8.1 經檢驗凡屬于下列情況之一者為不合格產品;
8.1.1 制作凈水器的材料不符合5.1的要求者為不合格產品。
8.1.2 凈水器的制作、裝配,外觀不符合5.2、5.3及5.4的要求者為不合格產品。
8.1.3 水壓力試驗不合格的產品為不合格產品。
8.1.4 凈水器在相對總產水量到達前發生下列情況之一者即為不合格產品:
a.包括GB5749感官性狀和一般化學指標中的色度,渾濁度,揮發酚,陽離子合成洗滌劑4項指標;毒理學指標所含15個指標;放射性指標中所含2個指標,亞硝酸鹽氮;以及采用三碘樹脂時出水中的碘化物;本標準表2所列凈水效率1~3項指標在內的一共26項指標中A級產品同時發生2項不符合各自標準的要求,B級產品同時發生4項不符合各自標準的要求;
b.帶有消毒裝置或措施的供生飲的凈水器,細菌總數或總大腸菌群有一項不符合GB5749的要求;
c.Ames致突變試驗不符合本標準表2的要求。
9 標志、包裝、運輸和貯存
9.1 每只凈水器應在規定的位置上固定銘牌,銘牌的內容規定如下:
a.制造廠名及商標;
b.產品名稱及型號標記;
c.產品制造編號(或日期)或生產批號;
d.產品的主要參數—活性炭體積、產水率、額定產水率、額定總產水量;
e.工作壓力;
f.質量等級標志;
g.有無消毒裝置。
9.2 包裝
9.2.1 包裝方法:家用凈水器用硬紙箱包裝;集團用凈水器視其重量決定用硬紙箱包裝或木箱包裝。
9.2.2 包裝應防潮、防震、凈水器進水和出水口應有防護套,包裝件外形尺寸應符合國內外運輸方法的有關規定,包裝頂部宜為平頂。
9.2.3 產品裝箱前應使其重心位置居中靠下,重心偏高的產品應盡可能采用臥式包裝,重心偏離中心較明顯的產品應采取相應的平衡措施。
9.2.4 包裝箱應有足夠的強度,起吊試驗,堆垛試驗和公路運輸試驗,應符合有關的規定。
9.2.5 產品應進行防雨包裝,在箱內采取必要的防雨措施,可在產品外部罩塑料罩,包裝箱頂蓋應采用雙層防水材料。
9.2.6 包裝標志應使用沖洗不掉的油漆、油墨等準確、清晰、牢固地噴刷或印刷在箱面上,其標志包括:
a.產品型號、名稱、規格和數量;
b.箱號;
c.箱體最大外形尺寸L×b×h(mm);
d.凈重與毛重(kg);
e.中華人民共和國制造(國內發運不需如此標志)。
9.2.7 產品分多箱包裝時,箱號應采用分數表示,分子為箱號、分母為總箱數,主件箱應為1號箱。
9.2.8 凡需起吊的和重心明顯偏離中心的包裝件,應標注“由此起吊”和“重心”的標志,并準確噴刷在包裝件相應的位置上。
9.2.9 隨帶文件
a.使用說明書
說明使用方法,注意事項,裝入的活性炭體積,亞硝酸鹽氮控制(大于0.03mg/L時應清洗活性炭,活性炭清洗或更換條件及更換方法),不帶消毒裝置的凈水器必須說明出水不能生飲;
b.產品合格證;
c.裝箱單;
d.備附件清單;
e.其他有關技術資料。
分離包裝時,隨帶文件應放在主件箱內。
附錄A 凈水器用活性炭半脫氯值測定方法(補充件)
A1 原理
在給定條件下,含氯水通過活性炭層后,出水中余氯濃度恰好等于進水中余氯濃度1/2時所需要的炭層高度,被稱為活性炭的半脫氯值,以“cm”表示。
設在給定的流速下,余氯濃度為C0的水通過高度為L的炭層后,出水濃度為C,則有:
1og=C0/C=KL (A1)
式中 系數K與水的流速u有關。而實驗表明,u與L有如下關系:
 (A2) (A2)
式中 h為半脫氯值。
為求K值,由A1、A2式得:
K=1ogC0/C/hu (A3)
根據半脫氯值定義,令C=1/2C0,且令u=1cm/s
則有:
K=log2/1/h=0.301/h (A4)
代入A1式得
logC0/C= 0.301/h L (A5)
即: h=0.301L /logC0-logC (A6)
通過(A6)式,即可在水流速度為1cm/s的條件下,從已知裝炭高度L和進出水濃度C0及C,計算出半脫氯值h。
A2 儀器和試劑
A2.1 儀器
a.水槽 501。
b.測定管 截面積3.14±0.26cm2,結構及尺寸見GB 7702《煤炭顆粒活性炭—有效防護時間測定總方法》附錄A《防護時間測定管及其校正方法》。
c.轉子流量計 LZB-6型,2.5~25L/n。
d.振蕩器 振幅≤2mm,頻率10~25Hz。
e.pH酸度計
f..溫度計 100°C
g.試驗篩 篩孔1.0和2.5mm
h.秒表
i.微量滴定管 5mL
j.容量瓶 200mL和500mL
k.研缽
l.燒杯 250mL
m.三角瓶 500mL和1000mL
A2.2 試劑
a.碘化鉀(分析純)
b.氫氧化鈉(分析純)預先配成10%溶液
c.鹽酸(分析純)預先配成5%溶液
d.乙酸(分析純)
e.次氯酸鈉溶液(化學純)
f.淀粉液1%
g.硫代硫酸鈉標準液1/2Na2S2O3的濃度0.01ml/L
A3 測試準備工作
A3.1 試驗條件
試驗氯水濃度 C=5.0±0.5mg/L
水的流速 u=36m/h(1cm/s)
裝炭高度 L=1.0±0.1cm
A3.2 測試裝置(見圖A1)
A3.3 試驗用氯水的制備
A3.3.1 分析次氯酸溶液原始濃度
A3.3.1.1 量取次氯酸鈉溶液2mL于200mL容量瓶中,用蒸餾水稀釋至刻線。
A3.3.1.2 將上述稀釋液傾入500mL三角瓶中,加碘化鉀約0.5g,再加10mL乙酸。
A3.3.1.3 用硫代硫酸鈉標準溶液滴定至淡黃色,加入淀粉液約1mL并繼續滴定至藍色消失為止。
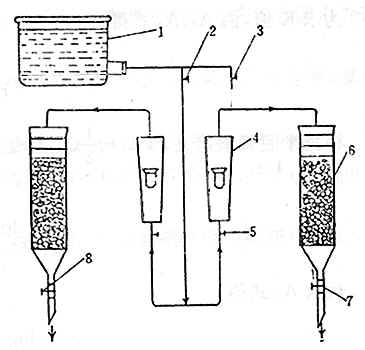
圖A 半脫氯值試驗裝置圖
圖中:1——水槽;2、3、7、8——活塞(或夾子);4——轉子流量計;5——調節閥門;6——測定管
A3.3.1.4 按A7式計算次氯酸鈉溶液濃度。
C=N1u1×35.45×1000/2 ×mg/L (A7)
式中 N1——硫代硫酸鈉溶液1/2Na2S2O3的濃度,mol/L;
u1——硫代硫酸鈉溶液滴定毫升數,mL;
C——次氯酸鈉溶液濃度,mg/L;
35.45——氯(C1或C1-)的摩爾質量,g/mol。
A3.3.2 配制氯水
A3.3.2.1 在水槽中加入足夠(15L以上)蒸餾水,水溫保持在20±3°C,再按氯水濃度要求加入適當的次氯酸鈉溶液,并用鹽酸和氫氧化鈉溶液調節其pH值至7.0~7.5。
A3.3.2.2 取200mL氯水按A3.3.1.2和A3.3.1.3所述步驟進行滴定,并按A8式計算其濃度。氯水濃度必須調節到5.0±0.5mg/L 。
C=N1u1×35.45×1000/2 ×mg/L (A8)
式中 C——所配氯水濃度,mg/L;
N1、u1等,意義同A7式。
A3.4 炭樣準備
將炭樣在研缽中破碎,用試驗篩手工篩取1.0~2.5mm篩份約100mL,置于250mL燒杯中,加蒸餾水至炭面以上1cm,煮沸10~15min,冷卻后去掉炭粉備用。
對于顆粒尺寸小于2.5mm的炭樣,不需破碎及篩選即可直接攝取,經處后備用。
A3.5 轉子流量計和測定管校正
A3.5.1 分別求出轉子在不同高度時的流量,繪出轉子高度與流量關系曲線。
A3.5.2 按GB 7702附錄A規定的方法測出測定管的內徑。
A3.5.3 計算出流量為36m/h(1cm/s)時轉子的高度,并標示在流量計上。
A4 測定步驟
A4.1 裝炭樣,將測定管下端7關嚴(或夾緊),先注入適量的蒸餾水(以裝好炭樣后水面高出炭面2~3mm為宜)管內得有氣泡。然后豎直于振蕩器上。邊振蕩邊分批加入準備好的炭樣,裝炭高度為10±0.1cm。炭層裝緊密(振蕩中炭層顆粒排列的整齊,高度不再變化)后才可停止振蕩,炭層內不得有氣泡,否則應重新裝炭。
A4.2 將裝好炭樣的測定管按圖接好,檢查活塞2和調節閥門5是否關嚴。
A4.3 開啟7(這時不應有水流出),再開啟2,然后同時打開兩個調節閥門5和開啟秒表記時,迅速調節好流量。
A4.4 試驗開始30min后,用容量瓶分別收集500mL流出液(28min時收集流出液預先洗滌容量瓶),按3.3.1.2和3.3.1.3所述步驟進行滴定,并按A9式計算出水濃度C,同時從水槽中取樣200mL測定進水濃度C0。
C=N1u1×35.45×1000/500 ×mg/L (A9)
(方法同A3.3.2.2)。
A4.5 根據A6式計算炭樣的半脫氯值,每個樣品測定兩個結果,平行誤差不得超過1cm,取其平均值。
A5 注意事項及說明
A5.1 試驗氯水濃度必須控制在5.0±0.5mg/L,由于次氯酸易揮發,配制試驗氯水時可適當偏上限。
A5.2 試劑的配制標定和使用期限按GB 601。
附錄B 凈水器用活性炭pH,氯化物、砷、鋅、鎘、鉛測定方法(補充件)
B1 適用范圍
本標準為凈水器用活性炭的檢驗方法。
B2 采樣方法
每生產批號,生產年月日及其生產單位,任意取兩個以上容器,并分別采集能盡量代表整體的必要量的試樣,保存在不受外界氣體影響的氣密性容器中。
B3 檢驗項目
pH值,氯化物,砷,鋅,鎘,鉛含量。
B4檢驗方法
B4.1 一般事項
B4.1.1 化學分析中的用水,在本檢驗方法中為蒸餾水的去離子水,其導電率為2us/cm以下。
B4.1.2 用未干燥試樣進行試驗,預先將部分未干燥試樣按B4.2求出干燥減量。利用該值將未干燥試樣量換算為試樣干燥重量。
B4.1.3 一般檢驗用溶液,按下述方法制備水萃取液和酸萃取液。
B4.1.3.1 水萃取液,準確稱取5g試樣(換算至干燥重量),將準確量取的500mL去郭子水放入1000mL帶塞三角瓶中。用磁攪拌器攪拌30min,用5號濾紙(直徑11cm)過濾,棄去初濾液30mL,其余濾液為水萃取液。
B4.1.3.2 酸萃取液,準確稱取5g試樣(換算至干燥重),預先將10N硝酸加入精制水中,調整pH值為4~5,準確量取500mL溶液,傾入1000mL燒杯中,放入稱好的試樣,保持微沸10min,冷卻后加水至加熱前量,用干燥的5號濾紙(直徑11cm)過濾,棄去初濾液30mL,余下的用作測定。
B4.2 干燥減量
本方法為測定試樣在110~120°C下干燥2h的減量。
B4.2.1 儀器:扁形稱量瓶40mm。
B4.2.2 檢驗操作
稱取2g試樣于已知重量的扁形稱量瓶底部,其厚度盡可能均勻,取下瓶蓋將稱量瓶和蓋子在110~120°C干燥箱中干燥2h,然后置于干燥器中冷卻,求出減量g數(a),按B1式計算干燥減量%。
干燥減量=a(g)/試樣(g)×100% (B1)
B4.3 pH值
本法為測定試樣水萃取液pH值。
B4.3.1 儀器
pHs-3 酸度計
B4.3.2 檢驗溶液:采用B4.1.3中水萃取液。
B4.3.3 檢驗操作:將試驗溶液在20°C條件下用酸度計測定。
B4.4 氯化物
本法所采取的試樣水萃取液是以鉻酸鉀溶液作指示劑,用硝酸銀溶液滴定,求出氯化物含量。
B4.4.1 試劑
(1)鉻酸鉀溶液:將5g鉻酸鉀溶于少量去離子水中,加5%硝酸銀溶液至生成少量紅色沉淀物,然后過濾,其濾液加去離子水100mL。
(2)0.1mol/L氯化鈉溶液:預先在500~600°C干燥40~50min,在硫酸干燥器中冷卻。稱取5.844g氯化鈉于1000mL容量瓶中加去離子水至刻度。
(3)0.01mol/L氯化鈉溶液:量取100mL0.1mol/L氯化鈉溶液于1000mL容量瓶中。加去離子去至刻度。
(4)0.01mol/L硝酸銀溶液:稱取1.7g硝酸銀于1000mL棕色容量瓶中加去離子水至刻度。
標定系數:吸取25mL0.01mol/L氯化鈉溶液于三角瓶中,加0.2mL鉻酸鉀溶液,用0.01mol/L硝酸銀溶液滴定至出現淡黃棕色不再消失,求出所需的0.01mol/L硝酸銀溶液mL數(a),按B2式計算標定系數:
標定系數-(F)=25/a (B2)
式中 F——0.01mol/L硝酸銀溶液標定系數。
本溶液1mL相當于0.355mg氯化物。
B4.4.2 儀器:三角瓶。
B4.4.3 檢驗溶液,采用B4.1.3試樣水萃取液。
B4.4.4 檢驗操作
吸取50mL檢驗溶液于三角瓶中,pH值后(1),加0.2mL鉻酸鉀溶液,用玻璃棒邊攪拌邊加數滴0.01mol/L硝酸銀溶液至出現淡黃褐色(2)。求出所需0.01mol/L硝酸銀溶液mL數(a),按B3式計算氯化物%:
氯化物%=aF×0.355×0.01/試樣(g)×50/500×100 (B3)
式中 F——0.01mol/L硝酸銀溶液標定系數。
注:(1)檢驗溶液為酸性時用0.02mol/L氫氧化鈉中和,堿性時用2.02mol/L硫酸中和。
(2)反應終點時,在另一同型三角瓶中取50mL去離子水,最好與這時所加的0.2mL鉻酸鉀溶液的顏色進行比較來判斷。
B4.5 砷含量的測定
本法將試樣酸萃取液中的砷制成砷化氫,用二乙氨基二硫代甲酸銀吡啶溶液吸收時呈橙黃色—紅色,與同法處理得到的標準液的顏色相比較,求出砷含量。
B4.5.1 試劑
(1)氯化亞錫溶液;稱取40g氯化亞錫(2個結晶水,不含砷)溶于100mL鹽酸中,該溶液在使用前配制。
(2)碘化鉀溶液:稱取15g碘化鉀溶于100mL去離子水中,該溶液在使用前配制。
(3)不含砷鋅粒。
(4)砷標準液
預先將三氧化二砷(標準試劑)在乳缽中研細,在105~110°C恒溫干燥箱中干燥3~4h,置于硫酸干燥器中,冷卻后從中稱取0.132g于500mL燒杯中,加5mL5mol/L氫化鈉溶液溶解,加400mL精制水,而后再加1mol/L硫酸進行中和(用石蕊試紙鑒定)后稱入1000mL容量瓶中,用少量去離子水沖洗燒杯,將沖洗液并入容量瓶中加去離子水至刻度,制成砷貯存液。
吸取砷貯存液10mL于另一個1000mL容量瓶中,加10mL1mol/L硫酸后加去離子水至刻度。
1mL砷溶液含砷(As)1.0µg。
(5)乙酸鉛溶液:稱取10g已酸鉛(3個結晶水)加100mL去離子水和1滴6mol/L乙酸溶解,密閉保存。
(6)吸收溶液1稱取0.25g二乙氨基二硫代甲酸銀(C5H10NS2、Ag)研碎后用少量氯仿溶解,加入1.0mL三乙酸胺(C6H15NO3),再用氯仿稀釋到100mL,必要時,靜置后過濾至棕色瓶內,貯存于冰箱中。
B4.5.2 儀器
(1)砷檢驗裝置:由三角發生瓶、導管和吸收管組成。
(2)光度計:分光光度計。
(3)檢驗溶液:采用B4.1.3試樣酸萃取液。
(4)檢驗操作
吸取100mL檢驗溶液于瓷表面皿上,在水浴上蒸發濃縮到25mL,移入砷檢驗裝置發生瓶中,將蒸發皿用少量水沖洗,并將洗液并入發生瓶中,加5mL鹽酸,并加去離子水至40mL左右。再加2mL磺化鉀溶液靜置2~3min后加1mL氯化亞錫溶液混勻靜置15min左右。往發生瓶內加3g鋅粒,立即將發生瓶、有乙酸鉛棉花的導管和加入5mL二乙氨基二硫代甲酸銀溶液的吸收管連接起來。在常溫下產生氫氯1h,以吸收管中的溶液制成檢測液。
在幾個砷檢驗裝置發生瓶中分別吸取適量的(1~20mL)砷標準液,分別加入5mL鹽酸,加水至40mL,另外將5mL鹽酸加到另一砷檢驗裝置發生瓶中,加水40mL左右,以下分別與檢驗溶液進行同樣處理,將其制成標準液和空白試驗液。
將檢驗溶液、標準液和空白試驗液吸入比色管中,用分光光度計測定在波長540mm附近的吸光度,根據測得的檢驗溶液的吸光度在砷含量標準曲線上求出檢驗溶液中砷含量µg數(a),按B4式算出砷的µg/g值。
砷(As µg/g)=a/試樣(g)×100/500 (B4)
B4.6 鋅含量的測定
B4.6.1 吸光光度法,本法用試樣酸萃取液、在弱酸性中存在硫代硫鈉的情況下,用雙硫腙四氯化碳萃取鋅時,呈紫色,與同樣處理得到的標準液顏色相比較,求出鋅含量。
B4.6.1.1 試劑
(1)鋅標準溶液:稱取0.4398g(ZnSO4·7H2O)溶于去離子水中,加入10mL濃HCI,用去離子水在1000mL容量瓶中稀至刻度,搖勻,使用前取此溶液10.00mL,用去離子水在1000mL容量瓶中稀至刻度,搖勻。
此液1.0mL含1.00µg鋅。
(2)0.1%雙硫腙四氯化碳貯備溶液:稱取0.10g雙硫腙(C13H12N4S),在干燥的燒杯中用四氯化碳溶解后稀釋至100mL,倒入棕色瓶中,此溶液置冰箱內保存,可穩定數周。
如雙硫腙不純,可用下述方法純化:稱取0.20g雙硫腙,溶于100mL氯仿,經脫脂棉過濾于250mL分液漏斗中,每次用20mL3+97稀氨水連續反萃取數次,直至氯仿相幾乎無綠色為止,合并水相至另一分液漏斗。每次用四氯化碳10mL振蕩洗滌水相兩次,棄去四氯化碳相。水相用1+9硫酸溶液酸化至有雙硫腙析出,再每次用100mL四氯化碳萃取兩次,合并四氯化碳相,倒入棕色瓶中。置冰箱內保存。
(3)雙硫腙四氯化碳溶液,臨用前,吸取適量雙硫腙四氯化碳腙貯備溶液(2),用四氯化碳稀約30倍,至吸光度為0.4(波長535nm,1cm比色皿)。
(4)乙酸—乙酸鈉緩沖溶液(pH4.7),稱取68g乙酸鈉(NaC2H3O2、3H2O),用去離子水溶解后稀釋至250mL,另量取冰乙酸31mL,用純水稀釋至250mL,將上述兩種溶液等體積混合。
如試劑不純,將上述混合液置于分液漏斗中,每次用10mL雙硫腙四氯化碳溶液(3)萃取,直至四氯化碳相呈綠色為止,棄去四氯化碳相;向水相中加入10mL四氯化碳,振搖洗滌水相,棄去四氯化碳相。如此反復數次,至四氯化碳相不顯綠色為止,用濾紙過濾水相于試劑瓶內。
(5)25%硫代硫酸鈉溶液,稱取25g硫代硫酸鈉(Na2S2O3、5H2O),溶于100mL去離子水中,如試劑不純,按(4)所述方法純化。
(6)0.1%甲基紅指示劑,稱取0.1g甲基紅(C15H15N3O2),用60mL95%乙醇溶解后,加純水至100mL。
(7)1+1氨水溶液。
(8)1+7乙酸溶液,將10mL冰乙酸溶于70mL去離子水中。
(9)四氯化碳。
B4.6.1.2 儀器
所用玻璃儀器均需用1+1硝酸洗滌,然后再用不含鋅的去離子水沖洗干凈,不得用自來水沖洗。
(1)60mL分液漏斗。
(2)10mL比色管。
(3)分光光度計。
B4.6.1.3 檢驗溶液,用B4.1.3試樣酸萃取液。
B4.6.1.4 檢驗操作
吸取2.00mL檢驗溶液于60mL分液漏斗中,另取分液漏斗8個,依次加入鋅標準液1.0~8.0mL,向水樣與標準中各加去離子水至10mL,各滴甲基紅指示劑,用1+1氨水調節液剛顯黃色,再滴加乙酸溶液至紅色(pH約4.4),加5mL四氯碳,振蕩萃取甲基紅,棄去有機相,向各分液漏斗中加入5.0緩沖溶液混勻,再加入1.0mL硫代硫酸鈉溶液,混勻,再加入10.0mL雙硫腙四氯化碳溶液,強烈振蕩4min,靜置分層。
用脫指棉或卷細的濾紙擦去分液漏斗頸內的水,棄去最初2~3mL有機相,收集其后的有機相于干燥的10mL比色管內。
于535nm波長下,用1cm比色皿,以四氯化碳為參比,測定樣品標準液列溶液的吸光度。
繪制標準曲線,在曲線上求出鋅的µg量(a),按B5式計算鋅的µg/g含量:
鋅(Zn µg/g)=a/試樣×2/500 (B5)
B4.6.2 原子吸光光度法
本法將試樣酸萃取液稀釋,直接用原子吸光光度法求出鋅含量。
B4.6.2.1 試劑
鋅標準液:采用B4.6.1中的試劑。
B4.6.2.2 儀器
玻璃儀器:與B4.6.1中的相同。
原子吸收分光光度計。
B4.6.2.3 檢驗溶液:采用B4.1.3試樣酸萃取液。
B4.6.2.4 檢驗操作
吸取10mL檢驗溶液于100mL容量瓶中,加去離子水至刻度,作為檢驗溶液。
取適量的鋅標準液(1~10mL)于數個100mL容量瓶中,每只加10mL1M硝酸,再加去離子水至刻度,于另一100mL容量瓶中加入10mL 1M硝酸,加去離子水至刻度。
將檢驗溶液、標準液和空白試驗液用原子吸光光度計,使用鋅中空陰極管在波長213.8nm測定吸光度,根據所得到的檢驗溶液吸光度在鋅含量標準曲線上,求出檢驗溶液中鋅的µg含量(a),按B6式計算鋅的µg/g含量。
鋅(Zn µg/g)=a/試樣(g)×10/500 (B6)
B4.7 鎘含量的測定
B4.7.1 吸光光度法:本法用試樣酸萃取液,在堿性氰化鉀存在下,用雙硫腙三氯甲烷溶液萃取鎘,再用酒石酸溶液進行逆萃取,在堿性氰化鉀中,用雙硫腙三氯甲烷溶液再萃取時,呈淡紅色。與同樣處理到的標準液顏色相比較,求出鎘含量。
B4.7.1.1 試劑
(1)檸檬酸銨溶液,稱取40g檸檬酸銨溶解于去離子水中,并加去離子水至100mL刻度。
(2)酒石酸鉀鈉溶液:稱取250g酒石酸鉀鈉加去離子水溶解至1000mL刻度。
(3)氫氧化鈉、氰化鉀溶液(A):稱取400g氫氧化鈉和10g氰化鉀溶于去離子水中,直至1000mL刻度。置于聚乙烯瓶中貯存。本溶液保存期為1~2個月。
(4)氫氧化鈉、氰化鉀溶液(B):稱取400g氫氧化鈉和0.5g氰化鉀在去離子水中溶解,并加水至1000mL刻度。置于聚乙烯瓶中貯存。本溶液保存期為1~2個月。
(5)鹽酸羥胺溶液:稱取20g鹽酸羥胺加去離子水溶液,并加水至100mL刻度。
(6)精制三氯甲烷:加5%三氯甲烷容量的硫酸振蕩混勻,反復操作至硫酸層為無色。其次,再用20%(容量)的1mol/L氫氧化鈉溶液洗滌兩次,再用去離子水洗,然后加氧化鈣進行蒸餾。加入20%(容量)餾出液的0.5%鹽酸羥胺溶液振蕩混勻,待分層后,將三氯甲烷層用干燥的濾紙過濾。然后加1%(容量)的乙醇(分析純)置于棕色瓶中,放于冷暗處保存。
(7)濃雙硫腙三氯甲烷溶液:將雙硫腙在研缽中研成細粉,取100mg雙硫腙加1000mL精制三氯甲烷溶解,放于冷暗處保存。
(8)稀雙硫腙三氯甲烷溶液:取濃雙硫腙三氯甲烷溶液100mL,加精制三氯甲烷稀釋至1000mL刻度,置于棕色瓶中,放于冷處保存,在使用期間加溫至室溫。
(9)酒石酸溶液:稱取20g酒石酸在去離子水中溶解,并加至1000mL刻度,放于冷暗處保存。
(10)鎘標準液:稱取0.100g金屬鎘于燒杯中,加20mL去離子水和5mL鹽酸,并加2~3滴硝酸,在水浴上加熱溶解,并蒸發至干固。冷卻后加10mL鹽酸溶解移入1000mL容量瓶中,燒杯用去離子水洗凈,并將洗液并入容量瓶中,加去離子水1000mL至刻度,制成隔貯存液,置于聚乙烯瓶中貯存。使用時吸取鎘貯存液10mL于1000mL容量瓶中,加10mL鹽酸,再加去離子水至1000mL刻度。
1mL該溶液含鎘10µg。
B4.7.1.2 儀器
光度計:分光光度計。
玻璃儀器:與B4.6.2相同。
B4.7.1.3 檢驗溶液:用B4.1.3試樣酸萃取液。
B4.7.1.4 檢驗操作
吸取100mL檢驗溶液于200mL分液漏斗中,加6mL檸檬酸銨溶液混合均勻,加3mL酒石酸鉀鈉溶液,加15mL氫氧化鈉、氰化鉀(A)溶液和3mL鹽酸羥胺混后,再加15mL雙硫腙三氯甲烷振蕩1min靜置分層。
將分離的三氯甲烷層移入第2號裝有25mL酒石酸溶液的100mL分液漏斗中。在第1號分液漏斗中加10mL精制三氯甲烷振蕩1min靜置分層,將分離的三氯甲烷并入第2號分液漏斗中(1)。
注:(1)該操作應立即進行,第2號分液漏斗中不要加入水層。
將第2號分液漏斗振蕩2min后靜置分層,將分離的三氯甲烷層舍棄,再加5mL精制三氯甲烷振蕩1min,仍將三氯甲烷層舍棄。
在水層中加入0.3mL鹽酸羥胺溶液,并準確加入15mL稀雙硫腙三氯甲烷溶液和5mL氫氧化鈉、氰化鉀溶液(B)振蕩1min靜置,用鋪有脫指棉的漏斗過濾三氯甲烷,置于干燥的試管中,制成檢驗溶液。
取適量的鎘標準液(1~6mL)于200mL分液漏斗中,分別加去離子水100mL左右,在另一200mL分液漏斗中,加100mL去離子水,以下與檢驗溶液進行同樣處理所得到的三氯甲烷層,作為標準液和空白試驗液。
將檢驗溶液、標準液和空白試驗液移入比色皿中,用分光光度計測定波長518nm附近的吸光度,根據所得至的檢驗溶液的吸光度,在鎘含量標準曲線上,求出鎘µg含量(a),按B7式計算鎘的µg/g含量。
鎘(Ca µg/g)=a/試樣(g)×100/500 (B7)
B4.7.2 原子吸收分光光度法
本法將試樣酸萃取液蒸發濃縮,直接利用原子吸收分光光度法,求出鎘含量。
B4.7.2.1 試劑
鎘標準液,與B4.7.1.1(10)相同。
B4.7.2.2 儀器
原子吸收分光光度計。
玻璃儀器與B4.6.1相同。
B4.7.2.3 檢驗溶液,采用B4.1.3試樣酸萃取液。
B4.7.2.4 檢驗操作
準確吸取200mL檢驗溶液于瓷表面皿上,在水浴上蒸發濃縮至10mL,冷卻后加去離子水至總量為20mL,制成檢驗溶液。
取適量鎘標準液(2~10mL),分別加入幾個100mL容量瓶中,每瓶各加10mL1mol/L硝酸,加精制水至100mL刻度。另外,在100mL容量瓶中,取10mL1mol/L硝酸溶液,加去離子水至100mL刻度,制成標準液和空白試驗液。
將檢驗溶液、標準液和空白試驗液分別在原子吸收分光光度計上,用鎘中空陰極管測定波長228.8nm處的吸光度,根據檢驗溶液的吸光度在鎘含量標準曲線上,求出鎘µg含量(a),按B6式計算鎘的µg/g含量:
鎘(Cd µg/g)=a/試樣(g)×200/500 (B6)
B4.8 鉛含量的測定
B4.8.1 吸光光度法
試樣酸萃取液,在檸檬酸銨、鹽酸羥胺和氰化鉀存在下,用雙硫腙四氯化碳萃取鉛,再稀硝酸處理,再次從水中逆萃取四氯化碳層,在逆萃取液中加入氨性氰化鉀—檸檬酸溶液,用雙硫腙四氯化碳進一步萃取時所呈現的鮮紅色與同法處理所得到的標準液顏色相比較,求出鉛含量。
B4.8.1.1 試劑
(1)檸檬酸銨溶液:稱取50g檸檬酸銨,加100mL去離子水溶解,滴加10mol/L氨水使pH為8.5~9.0,調整pH時為放熱反應,因而在容器外部用水冷卻。
將上述配制的溶液移入200mL分液漏斗中,為除去含有不純物的重金屬,加入10mL雙硫腙四氯化碳貯存液振蕩混勻,靜置分層,并將四氯化碳層舍棄,反復操作至雙硫腙四氯化碳溶液保持原有的綠色為止。然后為除去過量的雙硫腙,加10mL精制四氯化碳振蕩混勻,反復進行這樣的操作至四氯化碳無色為止。
(2)鹽酸羥胺液:稱取20g鹽酸羥胺于燒杯中,加65mL去離子水溶解。滴加10mol/L氨水,在pH計上調整溶液pH為8.5~9.0。調整pH時為放熱反應,因而在容器外部用冰冷卻。
(3)氰化鉀溶液:稱取20g氰化鉀在去離子水中溶解,溶解后加去離水至200mL刻度。由于氰化鉀溶液不能進行精制,為調配其純度,吸取5mL氰化溶液于50mL帶塞比色管中,加5mL雙硫腙四氯化碳溶液振蕩混勻。如果雙硫腙四氯化碳層不呈現紫紅色即可使用,當呈現紫紅色時,則須用純的氰化鉀。
(4)氨性氰化鉀:檸檬酸溶液:在500mL氨水中加入10g氰化鉀、10g檸檬酸使之溶解,加去離子水至1000mL刻度。
由于該溶液不能進行精制,可用一部分溶液進行與氰化鉀溶液一樣的試驗。
(5)百里香酚藍指示劑:在研體中研細稱取0.1g百里香酚藍,加10.8mL0.01mol/L氫氧化鈉溶液,研磨至全部溶解后,移入250mL容量瓶中,加去離子水至250mL刻度。
(6)精制四氯化碳。
(7)雙硫腙四氯化碳貯存液:采用B4.6.1試劑。
(8)雙硫腙四氯化碳溶液:采用B4.6.1試劑。
(9)鉛標準液稱取0.2g硝酸鉛于1000mL容量瓶中,再加1mL硝酸于100mL去離子水中溶解,再加去離子水至1000mL刻度,作為鉛貯存液。使用時吸取該溶液10mL于200mL容量瓶中,加去離子水至刻200mL刻度。再從該溶液中吸取10mL于250mL容量瓶中加去離子水至刻度。
1.0mL該溶液含鉛0.25µg。
B4.8.1.2 儀器
光度計:分光光度計。
玻璃儀器:與B3.6.1相同。
B4.8.1.3 檢驗溶液:采用B4.1.3試樣酸萃取液。
B4.8.1.4 檢驗操作
吸取50mL檢驗溶液于100mL第1號分液漏斗中,再加10mL檸檬酸銨液和數滴百里香酚藍指示劑,直至溶液呈淡暗綠色為止,再滴加氨水(pH為8.5~9.0)。然后,在溶液中加2mL鹽酸羥胺溶液和4mL氰化鉀溶液,混合后再加5mL雙硫腙四氯化碳溶液,振蕩30s,靜置分層,將四氯化碳層移入100mL第2號分液漏斗中。在第1號分液漏斗中再加5mL雙硫腙四氯化碳溶液,振蕩30s靜置分層,將四氯化碳層并入第2號分液漏斗中。在第2號分液漏斗中加20mL0.1mol/L硝酸,激烈振蕩1min靜置分層,將分離的四氯化碳層舍棄。水層加去離子水50mL左右,然后加4mL氨性氰化鉀、檸檬酸溶液,再加5mL雙硫腙四氯化碳溶液,振蕩1min后,靜置分層。將四氯化碳層移入試管,將此作為檢驗溶液。
吸取適量的鉛標準液(1~20mL),分別放入數個100mL分液漏斗中,各加去離子水至總量為50mL。再取50mL去離子水放入另外100mL的分液漏斗中。分別與檢驗溶液進行同樣處理,在試管中取所得的四氯化碳層,將它作為標準液和空白試驗液。在比色皿中放入檢驗溶液、標準液和空白試驗液,用分光光度計測定波長510nm附近的吸光度,根據所得檢驗液的吸光度,在鉛含量標準曲線上,求出檢驗溶液的鉛µg量(a),按B9式計算鉛µg/g含量:
鉛(Pb µg/g)=a/試樣(g)×50/500 (B9)
B4.8.2 原子吸收分光光度法
本法將試樣酸萃取液蒸發濃縮,直接用原子吸光光度法,求出鉛含量。
(1)試劑
鉛標準液:在500mL容量瓶中加入300mL1mol/L硝酸,然后準確加入5mL鉛貯存液(B4.8.1試劑),再加1mol/L硝酸至500mL刻度,制成鉛標準液。
1mL該溶液含鉛1.25µg
(2)儀器
原子吸收光光度計。
玻璃儀器:與B4.6.1相同。
(3)檢驗溶液:采用B4.1.3試樣酸萃取液。
(4)檢驗操作:檢驗溶液按B4.7.2操作。
分別吸取適量的鉛標準液(2~50mL)放入數個100mL容量瓶中,各加10mL1mol/L硝酸,加去離子水至100mL刻度,另在100mL容量瓶中取10mL1mol/L硝酸加去離子水至100mL刻度。這些分別作為標準液和空白試驗液。
將檢驗溶液、標準液和空白試驗液在原子吸收分光光度計上用鉛中空陰極管測定波長283.3nm的吸光度,根據得到的檢驗溶液的吸光度,在鉛含量標準曲線上,求出檢驗溶液的鉛µg含量(a),按B10式計算鉛ppm含量:
鉛(µg/g)=a/試樣(g)×200/500 (B10)
附錄C 總溶解性有機碳檢驗方法(補充件)
水中溶解著許多不同有機物。在這些碳化合物中有些可以經生物或化學的過程進一步氧化,這些部分可以通過生化需氧量(BOD)及化學需氧量(COD)予以測定。但是,水中還存在著既不為BOD也不為COD測出的有機碳。總溶解性有機碳DOC的檢驗法則是可以測定水中所有溶解性有機碳的合適方法。本檢驗法采用燃燒—紅外法。
C1 應用范圍
C1.1 本法適用于測定生活飲用水中溶解性有機碳。
C1.2 本法最低檢測量為1mg/L。
C1.3 采集水樣,貯在玻璃瓶中,送化驗室立即測定,如不能即時測定,可將其保存在溫度為4°C的環境中,盡量不暴露在光和空氣中并盡量減少貯存時間。
C2 原理
將微量水樣在凈化的空氣流中注射至已加熱的填棄管中,水樣氣化,有機碳被氧化為CO2及H2O0用紅外分析器測量CO2,測定總碳和無機碳,用差減法計算總有機碳。
C3 試劑
C3.1 重蒸餾水:用無CO2重蒸餾水配制空白和標準溶液。
C3.2 DOC標準溶液:稱0.850g烘干的鄰苯二甲酸氫鉀(C6H5KO4)溶于CO2的重蒸餾水中,稀釋至1000mL,1mL=0.4mg碳。
C3.3 標準無機碳溶液:稱1.40g碳酸氫鈉和1.73g(于500~600°C烘干30min,取出在干燥皿中冷卻至室溫)碳酸鈉,稀至1000mL,1mL=0.4mg。
C4 儀器
總有機碳分析儀。
C5 測定步驟
C5.1 按照儀器操作要求,使儀器達到正常工作狀態。
C5.2 標準曲線制備。
C5.2.1 在DOC標準溶液中準確吸取0、5.0、10.0、15.0、20.0、25.0mL于100mL容量瓶中,稀釋成0、20.0、40.0、60.0、80.0、100.0mg/L的標準溶液系列。
C5.2.2 用微量注射器吸取20µL標準溶液,注射進樣,每只樣品重復三次,使峰高誤差不超過1刻度,取平均值。記錄標準及1份稀釋水空白的峰高。
C5.2.3 在無機碳標準溶液中準確吸取0、5.0、10.0、15.0、20.0、25.0mL于100mL容量瓶中,稀釋配制成0、20.0、40.0、60.0、80.0、100.0mL/L標準系列。
C5.2.4 用微量注射器吸取20µL標準溶液,注射進樣,每只樣品重復三次,使峰高誤差不超過一刻度,取平均值,記錄標準及一份稀釋水空白的峰高。
C5.3 樣品注射
C5.3.1 水樣預處理 水樣在離心管中經2000n/min離心機離心10min。
C5.3.2 用微量注射器吸取預處理后的水樣20µL注射進樣,每只水樣重復三次,使峰高誤差不超過一刻度,取平均值。
C5.4 計算
C5.4.1 從標準峰高減去空白值,按標準濃度與峰高制成的總碳與無機碳的回歸方程。
C總=a1h+K1 (C1)
C無=a2h+K2 (C2)
式中 C總——總碳濃度,mg/L;
h——峰高;
C無——無機碳濃度,mg/L;
K1和K2——常數;
a1和a2——常數。
C5.4.2 按C1(式)和C2(式)計算的總碳和無機碳兩者之差為總溶解性有機碳。
DOC=C總-C無 mg/L (C3)
C5.4.3 精密度
本法精密度為1~2%。
附錄D 水樣Ames致突變試驗方法(補充件)
D1 定義
Ames試驗系列用微生物的營養缺陷型突變株的回復突變性能來檢測環境致突變物,有些致突變物需經哺乳動物細胞代謝活化后,才通體現其致突變作用,Ames等人研究并提出了鼠傷寒沙門氏菌/哺乳動物肝微粒體試驗法,它是在動物體外將待測物質經肝微粒體酶系活化后,檢查其所誘發的沙門氏菌回變菌落數,即由不能自行合成組氨酸的營養缺陷型突變株(his-)回復為能自行合成組氨酸的(his+)菌落數。
D2 材料和試劑
D2.1 材料
D2.1.1 菌株 常用于水致突變試驗的鼠傷寒沙門氏菌采用兩種(Salmonella typhimurinm)組氨酸缺陷型菌株,即TA98,TA100,其中TA98用以檢出可引起移碼突變的致突變物,TA100用以檢出可引起堿基對置換突變的致突變物,凡用作試驗的菌株,均需經五項主要性狀的鑒定,通過后方可使用。
D2.1.2 XAD-2樹脂 將市售XAD-2樹脂在索氏抽提器中,用乙醚、丙酮、甲醇溶液分別回流8h,然后保存在甲醇液中備用。
D2.1.3 水樣濃縮 取水樣40L(混濁水須經過濾)以25~30mL/min的流量通過裝有10g洗凈的XAD-2樹脂的層析柱,濾畢用30mL丙酮,或30mL30%丙酮-甲醇混合液,以濾水時的同樣流量將吸附于樹脂上的有機物洗脫,洗脫液于45°C水浴上揮干,然后用二甲亞砜(DMSO)溶解定容至4mL,再倍比稀釋成每0.1mL含0.25L、0.5L、1.0L、水樣的三個濃度組備用。
D2.2 陽性致突變物
D2.2.1 甲基甲烷磺酸酯(MMS)、濃度0.2mL/mLDMSO溶液,于4°C冰箱保存,不穩定。
D2.2.2 2.7-二氨基芴(2.7AF)濃度20mg/mLDMSO溶液,于4°C冰箱保存,尚穩定。
D2.2.3 2-氨基芴(2AF)濃度2mg/mLDMSO溶液于4°C冰箱保存。
D2.3 肝微粒體酶(S-9上清液 )
D2.3.1 誘導 選成年雄性大鼠3只(每只體重約200g左右)按500mg/kg一次腹腔注射Aroclor1254(或國產多氯聯苯)注射液用玉米油配制濃度為200mg/mL,注射后第5天殺鼠,殺鼠前12h禁食(可飲水)。
D2.3.2 制備肝勻漿,大鼠斷頭處死,以無菌操作,打開腹腔,用冷凍的0.15MKC1溶液門脈灌注,摘出肝臟,稱重,用冷凍0.15MKC1溶液洗滌3次,以每克肝加3倍冷凍0.15MKC1溶液,放入組織搗碎機,20000n/min 1min,制成勻漿,將肝勻漿于低溫高速離心機9000g(11000n/minRPR-16)離心10min(0~4°C)取上清液為S-9組分,用小安瓿分裝,每瓶2mL,封口,以干冰丙酮凍結后,于液氮或-80°C低溫冷箱保存,備用,用時在室溫下解凍,剩余部分應丟棄,每批S-9應作活性檢驗。
D2.3.3 配制S-9混合液,每1mL S-9混合液中,含有表D1內容物。
表D1 S-9混合液的配制 | S-9上清液 | 0.1mL | | 0.4M MgCI2 | 0.02mL | | 1.65M MCI | 0.02mL | | IM 葡萄糖-61-磷酸 | 0.005mL | | 0.1M 輔酶 Ⅱ(NADP) | 0.04mL | | 0.2M磷酸鹽緩沖液(pH7.4) | 0.50mL | | 滅菌蒸餾水 | 補足總量為1mL | 可先將上述各溶液配制好,除菌后,放冰箱備用,上述各項操作中所用器皿、刀剪,溶液均需保持無菌,并在0~4°C下操作。
D2.4 培養基
D2.4.1 營養肉湯、牛肉膏0.5%,蛋白胨1%,氯化鈉0.5%調pH至7.2。分裝中試管,每管5mL,高壓滅菌備用。
D2.4.2 營養瓊脂,在D2.4.1營養肉湯中加瓊脂2%即成,按需分裝,滅菌備用。
D2.4.3 底層(基本養基)(即最低營養培養基)
D2.4.3.1 Vogel-Bonner(V-B)低限培養基E(50×)見表D2。
表D2 Vogel-Bonner(V-B)低限培養基E(50×)的配制 | 成 分 | 每 升 | | 溫蒸餾水(45°C) | 670mL | | 硫酸鎂(MgSO4·7H2O) | 10g | | 檸檬酸 | 100g | | 磷酸氫二鉀(K2HPO4)無水 | 500g | | 磷酸氫銨鈉(NaHNH4PO44H2O) | 175g | 用一只2L燒杯,用溫水加(熱)溫,使每種鹽完全溶解后,再加入下一種鹽,調整容量至1L,然后120°C20min滅菌,冷卻至室溫4°C冰箱保存。
D2.4.3.2 40%葡萄糖溶液 葡萄糖40g,加蒸餾水100mL112°C,10min滅菌。冷卻至室外溫,4°C冰箱保存。
D2.4.3.3 瓊脂15g,加蒸餾水930mL,120°C20min滅菌。
D2.4.3.4 將D2..3.1、D2.4.3.2、D2.4.3.3、趁熱混合,降溫到55°C左右時倒入無菌平皿(直徑9cm)每平皿250mL左右,此瓊脂平板可置室溫保存。
D2.4.4 表層培養基,瓊脂0.6%,氯化鈉0.5%,按所配表層培養基總量加入1/10體積的0.5m Mol L—組氨酸及0.5mMol生物素混合均勻后,趁熱分裝小試管,每管2mL,滅菌備用。
D3 致突變試驗方法
D3.1 試驗用菌液的準備 用無菌小木勺刮取TA98及TA100凍干菌種少許分別接種入5mL營養肉湯試管內。37°C水浴振蕩培養9~12h,活菌數在1~3×109/mL范圍供誘變試驗用,此菌液可在4°C冰箱內保存,供用一周,但不宜轉管接種。
D3.2 致突變試驗操作步驟 先在平皿蓋上做好標記,取融化了并保溫45°C的表層培養基一管(2nL),依次加入菌液0.1mL(需活化的加S-9混合液0.5mL),充分混合后迅速倒在底層培養基上(全部操作不要超過20s),平鋪待凝,注意避光,37°C培養48h,觀察結果,每一水樣濃度需平行接種三個平皿。
D3.3 對照 每次試驗均設置自發回變(即在表層培養基中不加待測物)及已知致突變物(TA100菌株加S-9混合液的用2AF濃度為20µg/10µL,不加S-9的用MMS濃度為2µL/10µL,TA98菌株加S-9用2AF,濃度為20µg/10µL,不加S-9的用2.7AF濃度為200µg/10µL)作為對照,以檢驗所用方法及步驟之可靠性,對照試驗亦需平行接種三只平皿。
D4 結果與評價
D4.1 結果 計數每一濃度三個平皿培養基上的回變菌落數的平均數,凡誘發回變的his+菌落數為自發回交his+菌落數的2倍以上時,并有劑量反應關系,即為致突變物,可以用突變率表示之。見式D1。
突變率=誘發回變his+菌落數/自發回變his+菌落數 (D1)
只有當突變率大于等于2.0時才為陽性,當待測物濃度達到1L水樣濃縮物/皿(1)時仍為陰性者,可報告為陰性結果。
注:(1)在一般試驗中當水樣濃縮物濃度達到1L/皿時不出現陽性反應,就定為該水樣致突變試實驗為陰性結果,但也可根據不同要求對水樣濃縮物濃度進行增減。
在觀察結果時,于平皿上his+回變菌落下必須有一層菌苔,作為背襯方可確認為his+回變菌落,該菌苔系his-菌利用表層培養基中所含之微量組氨酸生長分裂數次所形成,這種生長對產生誘變作用是必要的。
D5 菌株性狀鑒定
D5.1 試劑
D5.1.1 組氨酸/生物素培養基
無菌操作,組氨酸/生物素培養基的配制見表D3。
表D3 組氨酸/生物素培養基的配制 | 成 份 | 每 升 | | 瓊 脂 | 15g | | 蒸餾水 | 914mL | | 50×VB鹽溶液 | 20mL | | 40%葡萄糖 | 50mL | | 滅菌鹽酸組氨酸水溶液(每400mL 水含2g) | 10mL | | 滅菌0.5mM生物素 | 6mL | 分別高壓,將滅菌40%葡萄糖,v-B鹽和組氨酸加進熱的瓊脂溶液中,混勻,倒平板。
D5.1.2 氨芐青霉素培養基
無菌操作,氨芐青霉素培養基的配制見表D4。
表D4 氨芐青霉素培養基 | 成 份 | 每 升 | | 瓊 脂 | 15g | | 蒸餾水 | 914mL | | 50×VB鹽溶液 | 20mL | | 40%葡萄糖 | 50mL | | 滅菌鹽酸組氨酸水溶液(每400mL 水含2g) | 10mL | | 滅菌0.5mM生物素 | 6mL | | 無菌氨芐青霉素溶液(8mg/mL0.02M NaOH) | 3.15mL | 混勻,冷卻到大約50°C,再加入無菌生物素和氨芐青霉素溶液,混勻倒板。
D5.1.3 結晶紫溶液
無菌配制,稱取100mg結晶紫,溶于100mL滅菌蒸餾水中。
D5.1.4 15W紫外線燈
D5.2 菌株鑒定
幾株菌可在同一平皿上鑒定。
D5.2.1 組氨酸需要
用無菌棉拭子或金屬耳沾取少許各菌株培養液,然后平行劃線接種于組氨酸/生物素平皿,37°C培養24h,結果:應生長,在不含組氨酸的平皿上應不生長。
D5.2.2 深度粗糙(rfa)特性
先用滅菌棉拭子或金屬耳沾取少許各菌株培養液,平行劃線接種于營養瓊脂平皿,培養液干后,在平板中線與接種的垂直方向貼放6mm寬浸有結晶紫溶液液的濾紙條,37°C培養24h,結果:濾紙條周圍形成透明抑菌帶,直徑約14mm。
D5.2.3 △uvrB特性 紫外線敏感試驗
先用滅菌棉拭子沾取各菌株培養液少許,平行劃線接種于營養瓊脂平板,然后在垂直距平板33cm處用15W紫外線燈照射8s,照射時平板中線一側用黑紙板遮蓋,37°C培養24h,結果:紫外線照射部分無菌生長。
D5.2.4 R 因子鑒定 抗氨芐青霉素試驗
用無菌棉拭子沾取各菌株培養液少許,平行劃線接種于氨芐青霉素平板,37°C培養24h,結果:有R因子的在劃線處有菌生長;無R因子的,在劃線處無菌生長。
D5.2.5 自發回變鑒定
將0.1mL各菌株培養液加入2mL45°C的表層培養液中,充分混勻后倒入低限葡萄糖平板,37°C培養48h,作菌落計數結果,在不加S-9混合液的低限葡萄糖平板上,自發回變數TA98為35~50、TA100 120~200。
TA98、TA100是帶有R因子的菌株。
D6 S-9的活性檢驗
在每制備一批新的S-9組分時,為了確證其代謝活性,必須對其活性進行檢驗,可以用黃曲霉素B,或2-氨基芴等作陽性對照檢驗,結果表明應有良好的活性,才能使用。 附加說明:
本標準由建設部標準定額研究所提出。
本標準由建設部水處理設備器材標準技術歸口單位中國市政工程華北設計院歸口。
本標準由上海市自來水公司,上海市自來水公司給水設備工程公司(原節水設備總廠)負責起草。
本標準主要起草人:岳舜琳、張珍鈺、吳國平。
本標準委托上海市自來水公司負責解釋。
| 


